
Comprendre le diagnostic génétique
Des anomalies génétiques se situant au niveau du chromosome 15 sont à l’origine du Syndrome d’Angelman.
Le dénominateur commun est l’absence de contribution des gènes de la région 15q11-q12.
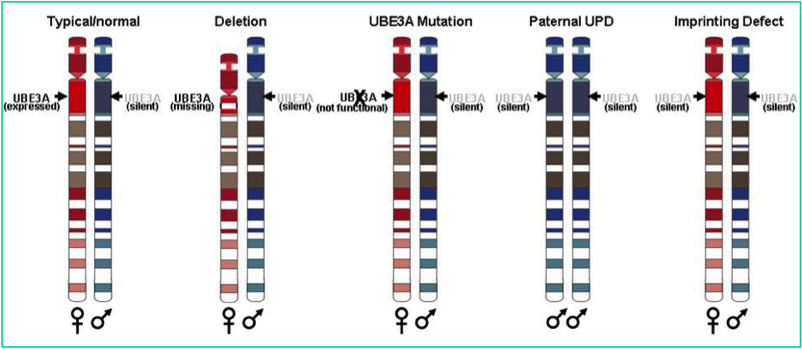
Quatre anomalies sont connues à ce jour et il reste environ 10% de patients pour lesquels aucune anomalie n’est décelable à ce jour.
Une microdélétion d’une taille d’environ 3 mégabases couvrant la région maternelle 15q11q12, est identifiée chez 66 % des patients.
Une mutation dans le gène UBE 3A (Ubiquitin protein ligase E3A), qui est l’anomalie de description plus récente. Ce gène localisé dans la région minimale critique 15q11q12 est décrit depuis seulement 1997 comme étant le gène du syndrome d’Angelman. La mutation en cause est retrouvée chez environ 16 % des patients. Cette anomalie est ainsi la deuxième anomalie la plus fréquente.Ces mécanismes moléculaires ont en commun l’absence de contribution physique ou fonctionnelle de la région q11q12 duchromosome 15 d’origine maternelle.
Méthylation : Une anomalie isolée de la région de régulation de l’empreinte du chromosome 15q11q12 maternel est environ retrouvée chez 4 % des patients.
Les gènes soumis à l’empreinte présents dans cette région ne s’expriment pas.
Une disomie uniparentale paternelle, en général il s’agit d’une isodisomie paternelle qui se définit par la présence d’un même chromosome 15 paternel dupliqué. Cette anomalie concerne 5 % des patients.
Aujourd’hui, il reste près de 9 % des sujets atteints du syndrome d’Angelman pour lesquels aucune des analyses actuellement disponibles ne met en évidence de défaut moléculaire.